
Earlier this year, my wife and I took a trip to the Grand Canyon in Arizona. From the top of the South Rim to the bottom of the canyon itself, we marveled at the majesty and grandeur of this natural wonder and the millions of years that it took to create it.
For me, the eons it took to produce the Grand Canyon is a metaphor for the biosimilar market.
We are now five years removed from the enactment of the Biologics Price Competition and Innovation Act of 2009 (BPCI). Since that time, the U.S. Food and Drug Administration (FDA) has successfully approved only one biosimilar and it is embroiled in litigation. While it feels like it may take tectonic forces to fully establish the biosimilar market on a grand scale, one that delivers meaningful savings to health systems and the patients they serve, some progress is being made.
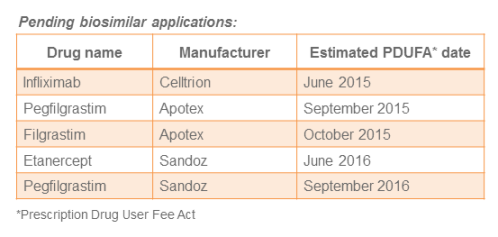
Pending applications
Five biosimilar applications are pending with the FDA as noted in the table below. Looking ahead to 2016, it looks like the FDA may begin scheduling advisory committee meetings. Such actions would signal progress towards approval of these pending biosimilars.

What’s in a name?
A lingering topic of debate between the branded industry and manufacturers pursuing biosimilar development is whether these agents should possess the same nonproprietary or “generic” name as the originator’s branded product. The FDA recently proposed guidance that the nonproprietary names of biologic products should be modified to include a four-letter suffix that has no meaning. According to the FDA, the purpose of this recommendation is to avoid inadvertent substitution of biosimilars not designated as interchangeable as well as supporting improved pharmacovigilance.
On the one hand, the FDA should receive credit for this recommended approach because it does not single out biosimilars, but instead applies to all biologics. On the other hand, the potential confusion and possible medication errors derived from a suffix, which is intentionally without meaning, would appear to be quite high as demonstrated in the table below.
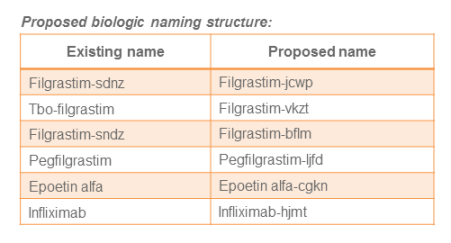
Many organizations have responded to the FDA encouraging the agency to alter its approach. However if this proposal does not change health care providers, particularly pharmacists, will need to determine how to ensure accurate prescription, documentation and reconciliation of biologic products throughout every aspect of patient care and across all health care information systems.
The “patent dance” and notification conundrum
As much uncertainty as exists with the regulatory aspect of biosimilars, the mystery surrounding key litigation issues may be even greater. Several key elements of the BPCI Act were poorly defined and thus have led to numerous lawsuits. This year saw the continuing saga of the Amgen vs. Sandoz litigation wind its way through district and federal courts. As of November, it appears that a final resolution of two primary issues may require intervention by the United States Supreme Court in 2016.
Along with fashioning the parameters by which the FDA can approve biosimilars, the BPCI Act also attempted to organize a methodology to expedite the process for litigating patent infringement issues. However due to the practicality of this process, it’s known affectionately as the “patent dance.” The process requires that within 20 days of the FDA accepting a biosimilar application, the manufacturer would be expected to share that information along with details of the biosimilar’s manufacture with the supplier of the originator, branded product.
The disclosure of proprietary information to the originator manufacturer has been met with less than an enthusiastic response by many potential biosimilar applicants. In addition, the lack of clarity of the language in the BPCI Act has prompted some biosimilar manufacturers to conclude this apparent requirement was in fact optional.
Another element of uncertainty concerns the notification requirement for biosimilar manufacturers. According to the BPCI Act, a biosimilar company is required to provide 180 days advanced notice to the originator company of the intent to market its biosimilar. Unfortunately, the statute does not state categorically how soon this notification can be provided. Notification prior to final approval, using an estimated decision date from the FDA, would allow a biosimilar manufacture to avoid any delay in launch.
However, as FDA approval dates are uncertain, delaying the notification until final approval would ensure that the intent to market would apply only to fully-licensed products. Not surprisingly, biosimilar manufacturers have advocated for the former interpretation and branded biologic suppliers have endorsed the latter approach.
Reimbursement question finally answered
Amidst the regulatory and legal uncertainty of biosimilars, another pertinent question received some clarification in 2015. The government had previously communicated its desire to encourage the use of biosimilars to drive cost savings through a reimbursement calculation different from that for generic medications. Under this approach, biosimilars are to be reimbursed at 100 percent of their average sales price (ASP) plus 6 percent of the ASP of the originator reference product. However, it was not known whether or not biosimilars of the same reference product would share J-codes. The Centers for Medicare & Medicare Services recently published its final rule stating that biosimilars of the same reference product will share a J-code. The ultimate impact this ruling will have on the total financial value of biosimilars remains to be determined.
Where does this leave us?
Ideally, we would have more biosimilars approved in the U.S. five years after the BPCI Act. Still, incremental progress is better than no advancement at all.
The approval of Zarxio showed that the FDA can utilize analytical data to substantiate biosimilar licensing including extrapolation across indications. In addition the naming guidance, while leaving a lot to be desired, does reflect the FDA’s attempt to navigate challenging issues. The migration of the Amgen vs. Sandoz lawsuit to the precipice of consideration by the Supreme Court suggests that a final conclusion to controversial aspects of a biosimilar’s pathway could be within sight.
If a vibrant biosimilars’ market is to become a reality in our country, pharmacists must be at the center of its implementation framework. Pharmacy practitioners will have to commit the time needed to support these products, provide the education required to prepare physicians and patients for these agents, and sustain the momentum of incremental progress until competition for the more frequently used biologics becomes a reality. These steps represent no small task, but are certainly achievable given a focused and sustained approach.
